

“When it comes to quality control, there’s always a way to improve. We believe in our solutions, and we stand behind them.”
Our growing list of certifications and approvals helps you meet the most demanding standards.
Shaping the future

OUR MISSION

DOWNLOADS
STANDARDS & COMPLIANCE
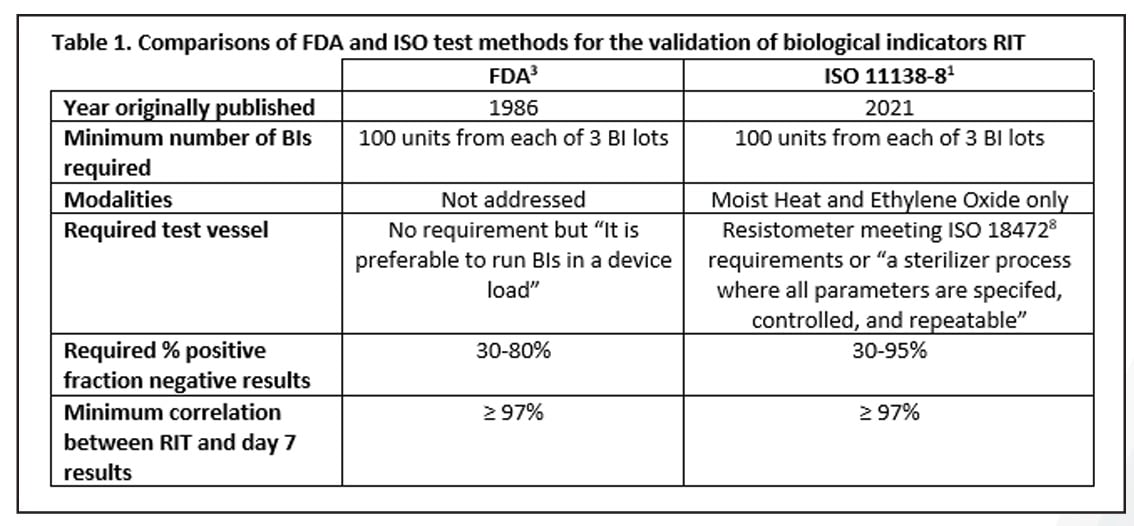
A Review of the Newly Released ISO 11138-8 Standard on the Validation of a Reduced Incubation Time for Biological Indicators
Read Mesa Labs' Spore News to learn about the newly developed ISO standard for the validation of a reduced incubation time as part of the 11138 series.

SOLUTIONS
The MeCo Solution: Improving Consistency, Accuracy and Robustness in SIP Validation
Read Mesa Labs’ Spore News to learn about how MeCo improves consistency, accuracy and robustness in sterilization in place (SIP) validation.

SERVICES