
By Kurt McCauley
[ Read in: Spanish | French | Italian ]
Mesa Labs manufactures its biological indicators (BIs) in accordance with relevant standards. A newly developed ISO standard for the validation of a reduced incubation time (RIT) for BIs has been published and is the subject of this Spore News. The standard has been designated as ISO 11138-81 and is the latest addition to the ISO 11138 series which is dedicated entirely to BIs. This standard is applicable to manufacturers of BIs as well as end-users who desire to establish or confirm an RIT.
Prior to the publication of ISO 11138-8, the only RIT method available was published by the Food and Drug Administration (FDA)2 in 1986, which has since been incorporated into an FDA guidance document titled “Biological Indicator (BI) Premarket 510(k) Submissions”3. This method was generally accepted globally and widely used for decades, however, in recent years several organizations outside of the United States (US) began rejecting the test method, in part because the method was specific to regulations in the US. To complicate things further, branches within the FDA itself (i.e., Center for Drug Evaluation and Research) no longer recognized the method (developed by the Center for Device and Radiological Health branch of the FDA), leaving many manufacturers of sterile product in a predicament. The rationale for rejecting this long-used method was never satisfactorily explained other than the FDA was unable to produce supporting evidence on the method development. As of this writing, the FDA method is still widely used, in large part because no alternate method had been presented, that is until July 2021, when the ISO 11138-8 method was published.
Working Group 4 (WG4) of ISO/TC 198, Sterilization of health care products--Biological Indicators, was responsible for the development of the RIT standard. Dr. John Gillis was a key figure in this effort and lead numerous studies resulting in several publications4,5,6. In April 2012, Schneider and Gillis7 provided a detailed history of the RIT project, which was initiated in 2000, delayed for several years, and reactivated in 2007. Through the 20+ years of the project’s lifetime, WG4 proposed, tested, and rejected numerous methods for RIT determination. As is often the case with standards development, rigorous debate ensued while developing the method. On one end of the spectrum, arguments were made that the last severely injured spore surviving the process should set the RIT. This was rejected as it was judged to be unnecessarily restrictive and set the RIT based on what most considered to be “outlier” data. On the other end of the spectrum, arguments were made that the RIT should be based on the last 10-100 severely injured spores. This method eliminated the need for hunting for the fraction negative exposure window which consumed much time and resources (one of the challenges with the FDA method). Rather, the exposure time was provided in a calculation that mathematically targeted low numbers of surviving spores; however, this method was judged to be too lenient, and it was difficult to correlate the calculated numbers of surviving spores with the empirically determined number of surviving spores.
As the project came to a close, the method that emerged from the years-long testing was somewhere in-between the two above mentioned options and was eerily similar to the FDA method. The commonalities between the FDA and ISO methods include the number of BIs required per test, targeting a fraction negative window, and a minimum correlation of 97% between the RIT and the 7-day results. The ISO method allows for a larger fraction negative window, but its method is restricted to moist heat and ethylene oxide modalities only. The ISO method requires the use of a resistometer8 or tightly controlled sterilizer for all testing while with the FDA method, this is not specifically addressed, although it is indicated that testing be conducted in a production sterilizer. A summary of these details is provided in Table 1.
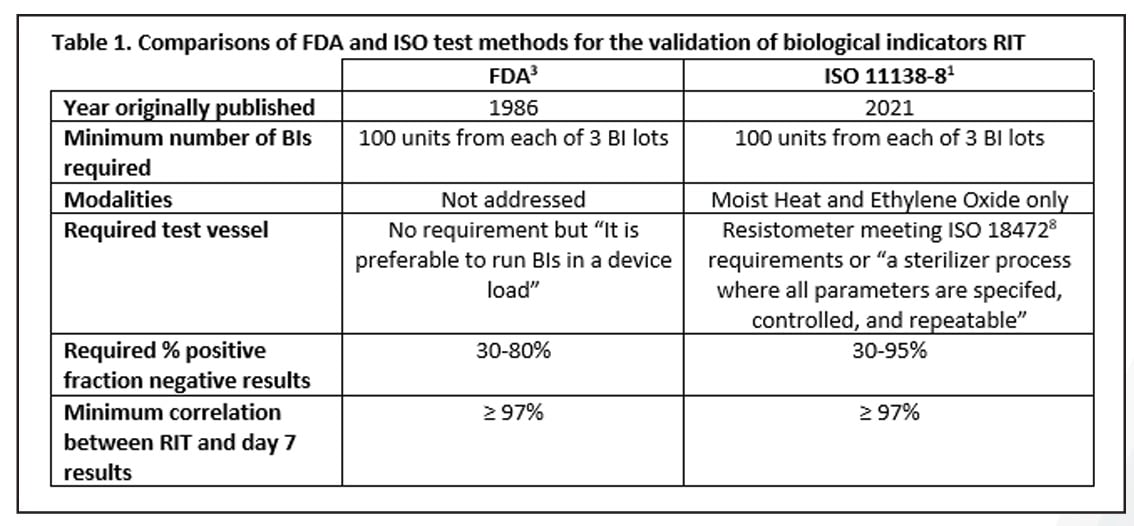
One limitation of the ISO 11138-8 standard (as noted above) is that it is only applicable with moist heat and ethylene oxide sterilization processes. What does this mean concerning the other sterilization modalities such as dry heat that have been successfully validated by the FDA method? The likely outcome will be that BI manufacturers will continue to validate these BIs per the FDA method and perhaps supplement their claims with data collected under the ISO validation method. After all, the two methods are nearly identical.
End users often wonder if it is a requirement that they too perform RIT studies on BIs used in their processes. ISO 11138-8 provides an answer for this, when it states, “This document is applicable to manufacturers of biological indicators (BIs) and to end users of BIs who intend to, if required by their quality system, establish, validate or confirm a RIT.”1 Additionally, the standard notes that it is not necessary that end-users conduct this test when it states, “The RIT validation for a spore strip that includes a recovery medium as a kit or a self-contained BI does not have to be repeated by the end user as long as the end user uses the BI with same sterilizing agent and BI incubation temperature as that used in the validation”1 .
With the publication of the ISO 11138-8 standard, an internationally developed and recognized test method for the validation of an RIT for biological indicators has been established.
There are four key principles noted by ISO in standards development; the standards, must respond to a need in the market, are based on global expert opinion, are developed through a multi-stakeholder process, and are based on a consensus.
The ISO 11138-8 standard meets all these criteria and is a welcomed addition to the ISO 11138-8 series.
[1] ISO 11138–8: 2021, Sterilization of health care products—Biological indicators—Part 8: Method of validation of a reduced incubation time for a biological indicator.
[2] FDA Guide for Validation of Biological Indicator Incubation Time (1986).
[3] Guidance for Industry and FDA Staff - Biological Indicator (BI) Premarket Notification [510(k)] Submissions, October 4, 2007.
[4] J.R. Gillis, G. A. Mosley, J.B. Kowalski, G. Krushefski, P.T. Nirgenau, K.J. McCauley, “Understanding Biological Indicator grow-out Times”, Pharmaceutical Technology, 34, (1), Jan 2010.
[5] J.R. Gillis, G. A. Mosley, J.B. Kowalski, G. Krushefski, P.T. Nirgenau, K.J. McCauley, P.M. Schneider “Understanding Biological Indicator Grow-Out Times--Part II”, Pharmaceutical Technology, Jun 2013.
[6] J.R. Gillis P. M. Schneider, G. A. Mosley, J.B. Kowalski, G. Krushefski, K.J. McCauley “Establishing a Minimum Incubation Time for Biological Indicators”, Pharmaceutical Technology, Dec 2013.
[7] P.M. Schneider, J.R. Gillis, “Biological Indicator Growout Times: The Impact on Establishing a Reduced Incubation Time Protocol”, Pharmaceutical Technology, Vol 2012 Supplement, Issue 3 April 2012.
[8] ISO 18472: 2018, Sterilization of health care products—Biological and chemical indicators—Test equipment.
SN071-V1