By Rick Gillis Ph.D., (Updated by Mesa Labs staff)
We are often asked by our customers if they need to confirm the reduced incubation time (RIT) for biological indicators (BIs). The US FDA RIT protocol (Guidance for Industry and FDA Staff, Biological Indicator (BI) Premarket Notification [510(k)] Submissions, Attachment II Examples of Validation of Biological Indicator Incubation Time) originated in 1986 and until 2021, was the only recognized protocol to reduce the incubation time for culturing biological indicators. A newly published standard, ISO 11138-8 Sterilization of health care products – Biological indicators – Part 8: Method for validation of a reduced incubation time for a biological indicator, provides an internationally recognized method for RIT validation, which is quite similar to the FDA RIT protocol. A brief history of the development of the new ISO standard was recently discussed in Spore News Vol. 15 No. 3, A Review of the Newly Released ISO 11138-8 Standard on the Validation of a Reduced Incubation Time for Biological Indicators, along with additional details on the method.
The intent of these protocols is for the manufacturer to determine the appropriate amount of time necessary to recover low numbers of injured spores, although certain provisions are made for end user testing as well. The manufacturer’s compliance with the protocol provides the end-user with confidence they can release product without fear of more biological indicators turning positive…the “what-if-we-missed-one” scenario. The RIT protocols are required for manufacturers of biological indicators who want to reduce the standard label claim to less than seven days. The seven-day timeframe has little scientific basis, whereas the US FDA and ISO RIT protocols are scientifically and statistically based.
Why do we as manufacturers want to reduce the incubation time? The answer is why not? Scientific advancements in sedimentation solutions have allowed manufacturers to produce spore crops with less vegetative debris, termed “clean spores”. Furthermore, advances in sporulation techniques have yielded more consistent resistance and germination. Advances in cultivation media have also assisted with faster germination. Therefore, due to the scientific advances in cultivation and sporulation media as well as post-processing of the spores, the obvious next step is to take advantage of these advances to achieve an RIT of less than seven days. The RIT allows end users to release sterilized product based on biological data in less than seven days. This results in shorter process times which reduce inventory costs, and allows faster turn-around time on critically needed medical supplies. In short, it allows the end user to get the critical biological information faster so that decisions to reprocess or release can be made faster. From a manufacturing standpoint, it allows the manufacturer to have shorter process times during manufacturing, faster release testing turn-around time, which means they can get customer orders completed more quickly.
To perform an RIT according to both the FDA and ISO methods, spores are injured during exposure in resistometers to sub-lethal times. One hundred biological indicators must be exposed per sublethal time. A fractional result must be obtained when cultured and the number of positive units required is slightly different per the two methods. The FDA method requires 30-80 positive units while the ISO method requires 30-95 positive units. The most probable number of spores per BI for this test is calculated using the Halvorson Ziegler equation of ln (n/r). n is the number of units tested (i.e., 100) and r is the number of units sterile. When 30 units are positive (per both the FDA and ISO methods), 70 units would be sterile. This would mean the most probable number of spores/BI = 0.36 spores. In the case of 80 units positive (per the FDA method), 20 units would be sterile, therefore, the most probable number of spores/BI = 1.61. In the case of 95 units positive (per the ISO method), 5 units would be sterile, therefore, the most probably number of spores/BI = 3. The RIT protocols are really analyzing the last surviving spore(s) in the BI, a very conservative test. The RIT for both methods is the time when 97% of the data available in 7 days of incubation is available to the analyst. The RIT time must be reproduced with three different lots of biological indicators. To get this type of control and reproducibility, every variable must be addressed. Consequently, performing an RIT is not trivial. It requires extensive control of critical experimental variables: the spores, the recovery medium, and the incubation conditions. The exposure conditions necessary to stress the BI to the last surviving spore must be precisely controlled.
What are the issues? There are three elements that can significantly influence the reduced incubation time results: (1) the spores – cultivation, cleaning, and preparation, which are all controlled by the manufacturer; (2) the media – highly specialized by the manufacturer to recover spores; (3) the incubator – only factor controlled by both the manufacturer and the user. Lack of control and consistency in any of these elements can lead to significant discrepancies in RIT.
The intent of the FDA and ISO methods is that the BI manufacturer establishes the RIT; however, there are situations where end-user testing may be required. Section 4.4 of the ISO standard states, “If an end user intends to use a RIT with a spore carrier/recovery medium combination or incubation conditions that have not been tested by a BI manufacturer (e.g. a spore strip used with a recovery medium not supplied as part of a kit), the end user shall establish the RIT for that system using the method provided in this standard.”
RIT testing can be performed in process vessels; however, achieving the proper fraction negative results is quite challenging. The physical dynamics of a process vessel are chaotic when compared to the resistometer used by BI manufacturers. The chamber size, the load, the precise delivery of the sterilant, and the precise expulsion of the sterilant are just a few examples of process variables that make it nearly impossible to use a process vessel for RIT. This array of process variables is impossible to control to the extent that reproducible sub-lethal exposures can be performed.
The intent of the RIT is to supply the end-user with the assurance that if the spores were not killed during the sterilization process, those spores will germinate and grow to yield a positive BI within the specified RIT. The RIT validation is designed to evaluate the ability to recover the last surviving spore. When cycles fail and yield a positive BI, it is a catastrophic failure; thousands of spores survive not just one or two spores. The reason this can be said is that all sterilization cycles have a test point when all BIs are killed. As shown in Figure 1, additional exposure time is added for sterility assurance.
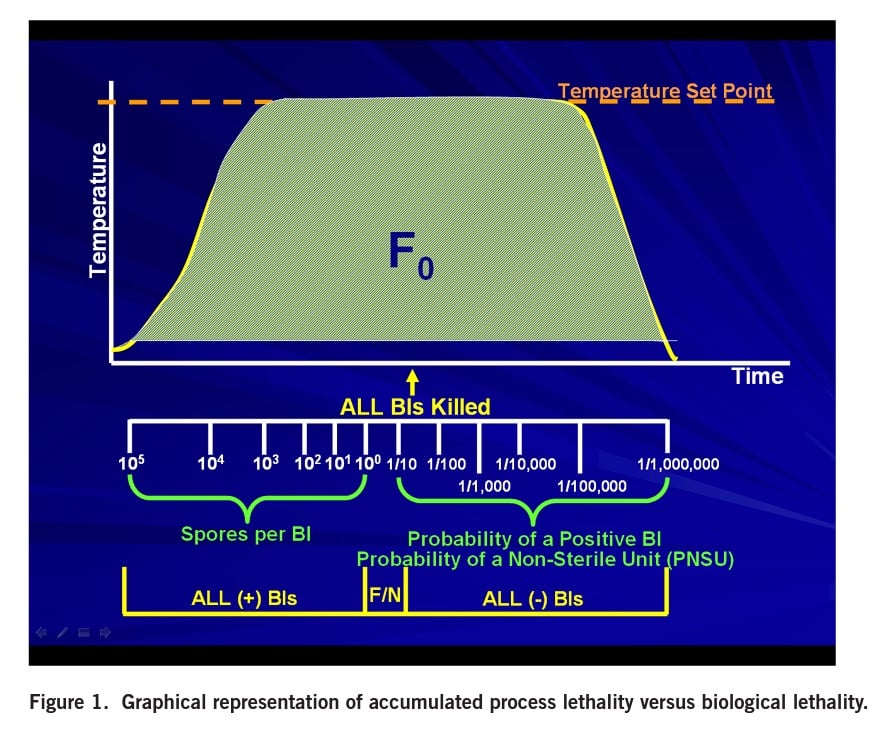
In order for a BI to survive, the cycle must lose all the lethality intended to be delivered in the sterility assurance portion. Typically, a BI failure occurs when recorded physical parameters are acceptable. Remember, it is the first positive biological indicator that fails the sterilization process, not the last. Customers will never see a sterilization failure with only one or two spores. It will be a catastrophic failure.
What would cause such a catastrophic failure? Validated cycles can fail for a multitude of reasons. These can include probe failure due to a calibration issue, a load configuration change causing air pockets, mechanical failure (i.e., steam traps, check valves), leaks, boiler issues, non-condensable gasses, poor vacuum, low ethylene oxide concentration, inadequate relative humidity, poor circulation, etc. (Figure 2).
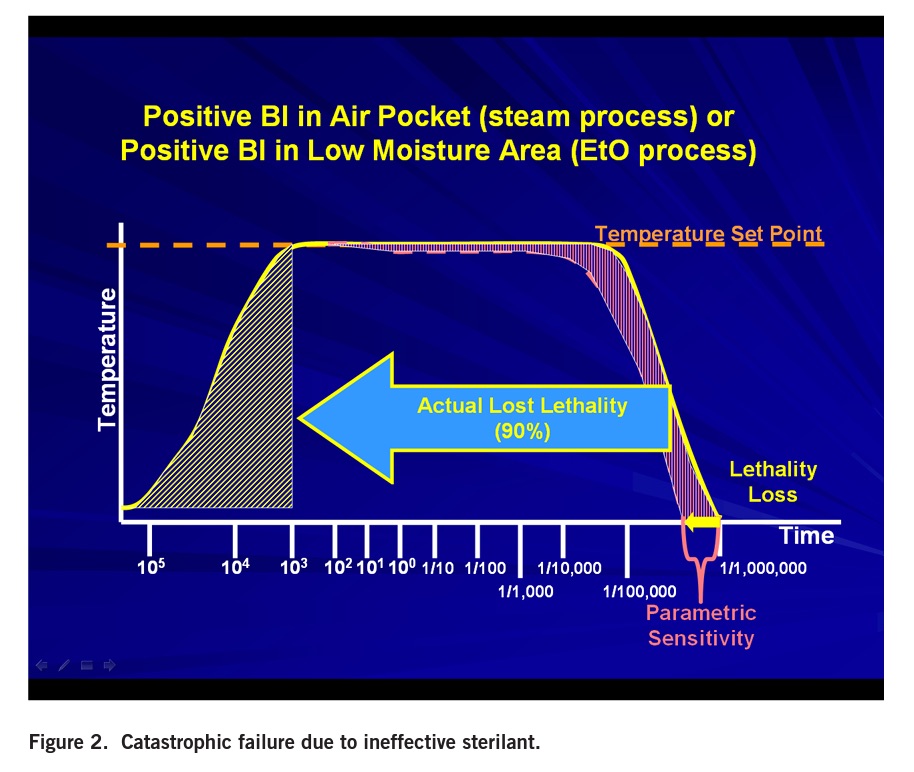
The impact of air pockets on steam efficacy in an autoclave can be significant. Dry heat (hot air) is 10 to 100 times less lethal than saturated steam. Air pockets can result from packaging barriers, (i.e., tortuous paths as found in catheters, lumens, etc.), and tightly packed loads.
Finally, although RIT is important to establish, it is primarily the manufacturer’s responsibility to do so. Customers (end users) will become very frustrated trying to verify the RIT in a process vessel. We at Mesa Labs go to great lengths to control our spores, our culture media, our incubators, and our resistometers to achieve the reproducible data necessary to establish the RIT. We welcome the opportunity to provide any pertinent information that regulatory departments may require.
We recommend that all users have temperature distribution studies of their incubators to ensure that their incubation conditions meet manufacturer specifications. The spores are killed by exposure to lethal conditions. The lethal mechanism is not sterilizer dependent. The rate of lethality can vary in sterilizers, but this test requires a lethal insult to reduce the spore challenge to approximately one spore… end of story.